

1
Esumi Y, et al.: Xenobiotica. 1994; 24(10): 957-964
2
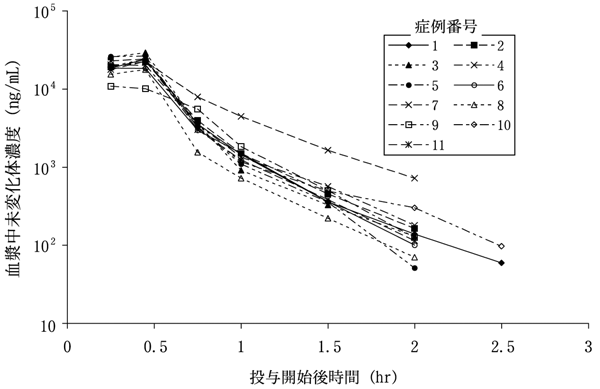
社内資料: 転移性乳癌患者におけるゲムシタビンとパクリタキセル併用投与時の薬物動態
3
福岡正博 他: 癌と化学療法. 1996; 23(13): 1825-1832
4
横山晶 他: 癌と化学療法. 1996; 23(12): 1681-1688
5
福岡正博 他: 癌と化学療法. 1996; 23(13): 1813-1824
6
Okada S, et al.: Japanese Journal of Clinical Oncology. 2001; 31(1): 7-12
7
Rothenberg M. L, et al.: Annals of Oncology.1996; 7(4): 347-353
8
Burris H. A, et al.: Journal of Clinical Oncology. 1997; 15(6): 2403-2413
9
Okusaka T, et al.: Cancer Chemotherapy and Pharmacology. 2006; 57(5): 647-653
10
Von der Maase H, et al.: Journal of Clinical Oncology. 2000; 18(17): 3068-3077
11
Albain K. S, et al.: Journal of Clinical Oncology. 2008; 26(24): 3950-3957
12
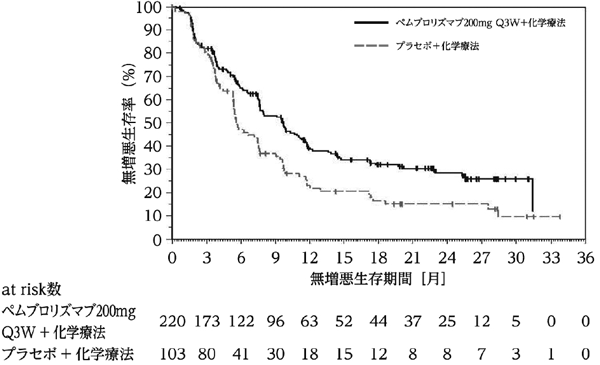
Cortes J, et al.: Lancet. 2020; 396: 1817-28
13
Plunkett W, et al.: Cancer Research. 1988; 48(14): 4024-4031
14
Plunkett W, et al.: Cancer Research. 1991; 51(22): 6110-6117
15
Plunkett W, et al.: Seminars in Oncology. 1995; 22(4 Suppl II): 19-25
16
Plunkett W, et al.: Purine and Pyrimidine Metabolism in Man VII. 1991; Part A: 125-130
17
Plunkett W, et al.: Molecular Pharmacology. 1990; 38(4): 567-572
18
Von Hoff D. D, et al.: Anti-Cancer Drugs. 1992; 3(2): 143-146
19
Peters G. J, et al.: Purine and Pyrimidine Metabolism in Man VII. 1991; Part A, 57-60
20
Bhalla K, et al.: Gynecologic Oncology. 1992; 45(1): 32-39
21
Momparler R. L, et al.: Anti-Cancer Drugs. 1991; 2(1): 49-55
22
Weber G, et al.: Biochemical and Biophysical Research Communications. 1992; 184(2): 551-559
23
Rockwell S, et al.: Oncology Research. 1992; 4(4-5): 151-155
24
Hertel L. W, et al.: Cancer Research. 1990; 50(14): 4417-4422
25
Plunkett W, et al.: Cancer Research. 1990; 50(12): 3675-3680
26
Braakhuis B. J. M, et al.: Cancer Research. 1991; 51(1): 211-214
27
Kristjansen P. E. G, et al.: Annals Oncology. 1993; 4(2): 157-160
28
藤田昌英 他: 癌と化学療法. 1994; 21(4): 517-523
29
Peters G. J, et al.: Seminars in Oncology. 1995; 22(4 Suppl II,): 72-79
30
Schultz R. M, et al.: Oncology Research. 1993; 5(6-7): 223-228


ゲムシタビン40mg相当量/mL(生理食塩液):用法・用量における溶解時最高濃度(ゲムシタビン200mg相当量/5mL生理食塩液又はゲムシタビン1g相当量/25mL生理食塩液)