









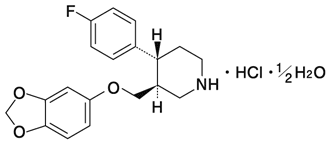
1
Chambers CD, et al.:N Engl J Med.2006;354:579-587
2
Kallen B, et al.:Pharmacoepidemiol Drug Saf.2008;17:801-806
3
Öhman R,et al.:J Clin Psychiatry.1999;60:519-523
4
入江 廣ほか:薬理と治療2000;28(Suppl 1):47-68
5
村崎光邦ほか:薬理と治療2000;28(Suppl 1):37-46
6
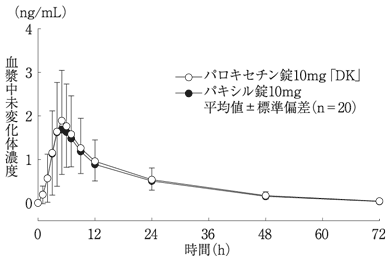
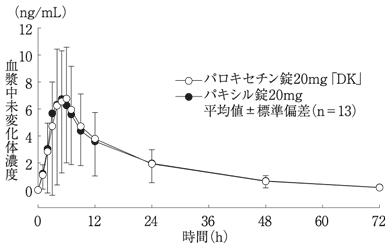
社内資料:生物学的同等性試験
7
20mg単回経口投与した時の食事の影響(パキシル錠:2000年9月22日承認、申請資料概要ヘ.3-7-1)
8
血球分配率(パキシル錠:2000年9月22日承認、申請資料概要ヘ.2-2-6)
9
Crewe HK,et al.:Br J Clin Pharmacol.1992;34:262-265
10
Özdemir V,et al.:Clin Pharmacol Ther.1997;62:334-347
11
Albers LJ,et al.:Psychiatry Res.1996;59:189-196
12
Hemeryck A,et al.:Clin Pharmacol Ther.2000;67:283-291
13
Sindrup SH,et al.:Clin Pharmacol Ther.1992;51:278-287
14
健常成人(西欧人)に14C標識塩酸パロキセチンを単回投与した時の吸収・代謝・排泄(パロキセチン錠:2000年9月22日承認、申請資料概要ヘ.3-5)
15
腎機能低下者(西欧人)に反復投与した時の体内動態(パキシル錠:2000年9月22日承認、申請資料概要ヘ.3-9-1)
16
Dalhoff K,et al.:Eur J Clin Pharmacol.1991;41:351-354
17
永田良一ほか:薬理と治療.2000;28(Suppl 1):89-110
18
三浦貞則ほか:薬理と治療.2000;28(Suppl 1):137-160
19
筒井末春ほか:薬理と治療.2000;28(Suppl 1):161-185
20
三浦貞則ほか:薬理と治療.2000;28(Suppl 1):187-210
21
三浦貞則ほか:薬理と治療.2000;28(Suppl 1):119-135
22
斎藤正己ほか:薬理と治療.2000;28(Suppl 1):211-223
23
片岡憲章ほか:薬理と治療.2000;28(Suppl 1):225-236
24
小林一広ほか:薬理と治療.2000;28(Suppl 1):237-252
25
筒井末春ほか:薬理と治療.2000;28(Suppl 1):271-294
26
筒井末春ほか:薬理と治療.2000;28(Suppl 1):295-314
27
筒井末春ほか:薬理と治療.2000;28(Suppl 1):253-269
28
上島国利ほか:薬理と治療.2004;32:577-591
29
第Ⅲ相試験(パキシル錠:2000年9月22日承認、審査報告書)
30
長期投与試験(パキシル錠:2000年9月22日承認、審査報告書)
31
Thomas DR,et al.:Psychopharmacology.1987;93:193-200
32
Gartside SE,et al.:Br J Pharmacol.1995;115:1064-1070
33
Lassen JB:Psychopharmacology.1978;57:151-153
34
Kennett GA,et al.:Neuropharmacology.1994;33:1581-1588
35
Perrault GH,et al.:Pharmacol Biochem Behav.1992;42:45-47
36
島田 瞭ほか:実中研・前臨床研究報.1996;20:163-167
37
Lightowler S,et al.:Pharmacol Biochem Behav.1994;49:281-285
38
抗不安作用(パキシル錠:2000年9月22日承認、申請資料概要ホ.1-1-2)
39
Cadogan AK,et al.:Br J Pharmacol.1992;107(Proc Suppl Oct):108P
40
非臨床試験に関する資料(パキシル錠:2000年9月22日承認、審査報告書)
41
非臨床に関する資料(パキシル錠:2000年9月22日承認、審査報告書)

