

1
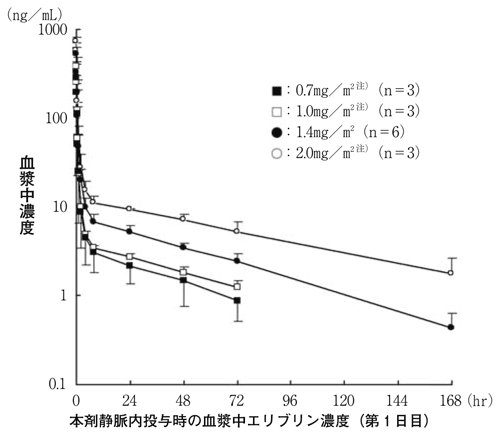
社内資料:日本人固形がん患者を対象とした臨床第Ⅰ相試験(国内試験)(2011年4月22日承認、CTD 2.7.6.3) [HAL-0050]
2
社内資料:エリブリンメシル酸塩の血漿たん白結合(2011年4月22日承認、CTD 2.6.4.4.3) [HAL-0051]
3
社内資料:14C-エリブリン酢酸塩由来放射能の分布(ラット、イヌ)(2011年4月22日承認、CTD 2.6.4.4.2.2) [HAL-0052]
4
社内資料:固形がん患者を対象としたマスバランス試験(外国試験)(2011年4月22日承認、CTD 2.7.6.4) [HAL-0053]
5
社内資料:エリブリンメシル酸塩のin vitro代謝(2011年4月22日承認、CTD 2.6.4.5.3) [HAL-0054]
6
社内資料:エリブリンメシル酸塩の母集団薬物動態解析(2011年4月22日承認、CTD 2.7.2.3.2.3) [HAL-0055]
7
Devriese L. A. et al.:Cancer Chemother. Pharmacol., 2012;70(6):823-832 [HAL-0168]
8
Tan A. R. et al.:Cancer Chemother. Pharmacol.,2015;76(5):1051-1061 [HAL-0638]
9
Devriese L. A. et al.:Invest. New Drugs, 2013;31(2):381-389 [HAL-0119]
10
Devriese L. A. et al.:Br. J. Clin. Pharmacol., 2013;75(2):507-515 [HAL-0135]
11
社内資料:進行又は再発乳癌を対象とした臨床第Ⅱ相試験(国内試験)(2011年4月22日承認、CTD 2.7.6.11) [HAL-0058]
12
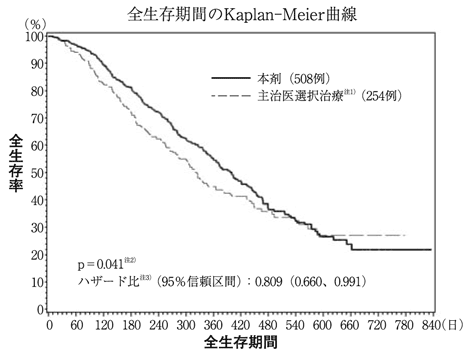
社内資料:進行又は再発乳癌を対象とした臨床第Ⅲ相試験(外国試験)(2011年4月22日承認、CTD 2.7.6.12) [HAL-0059]
13
社内資料:進行又は再発悪性軟部腫瘍を対象とした臨床第Ⅱ相試験(国内試験)(2016年2月29日承認、CTD 2.7.6.3) [HAL-0342]
14
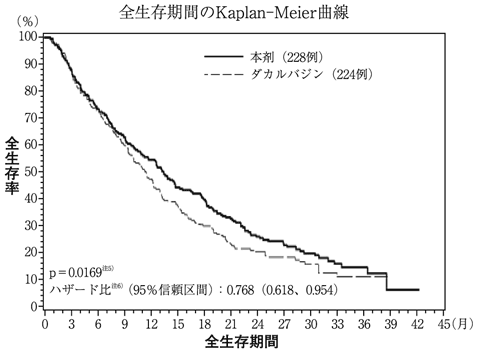
社内資料:進行又は再発悪性軟部腫瘍を対象とした臨床第Ⅲ相試験(外国試験)(2016年2月29日承認、CTD 2.7.6.1) [HAL-0343]
15
Towle M. J. et al.:Cancer Res., 2001;61(3):1013-1021 [HAL-0002]
16
Kuznetsov G. et al.:Cancer Res., 2004;64(16):5760-5766 [HAL-0003]
17
Jordan M. A. et al.:Mol. Cancer Ther., 2005;4(7):1086-1095 [HAL-0004]
18
社内資料:エリブリンメシル酸塩の作用機序に関する検討(2011年4月22日承認、CTD 2.6.2.2.1.2) [HAL-0060]
19
社内資料:エリブリンメシル酸塩のがん細胞増殖抑制作用(in vitro)(2011年4月22日承認、CTD 2.6.2.2.1.1) [HAL-0061]
20
社内資料:エリブリンメシル酸塩の抗腫瘍効果(in vivo)(2011年4月22日承認、CTD 2.6.2.2.2.1) [HAL-0062]
21
社内資料:エリブリンメシル酸塩と他の抗がん剤の併用効果(in vivo)(2011年4月22日承認、CTD 2.6.2.2.2.4) [HAL-0063]


(エリブリンメシル酸塩2.03mg相当量/100mL生理食塩液)