


1
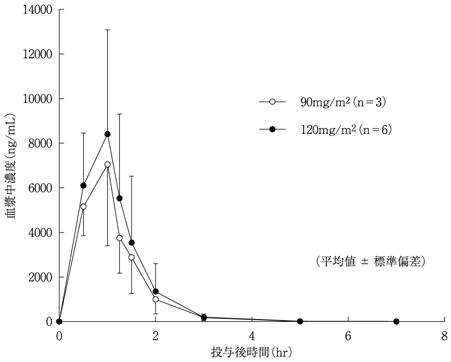
社内資料:薬物動態[国内第Ⅰ相臨床試験(2006001試験)]
(承認年月日:2010年10月27日、CTD 2.7.6.1)
(承認年月日:2010年10月27日、CTD 2.7.6.1)
2
社内資料:薬物動態[海外非臨床試験(KLG/06試験)]
(承認年月日:2010年10月27日、CTD 2.6.4.4.5)
(承認年月日:2010年10月27日、CTD 2.6.4.4.5)
3
社内資料:薬物動態[海外非臨床試験(DM-2008-006試験)]
(承認年月日:2010年10月27日、CTD 2.6.4.5.5)
(承認年月日:2010年10月27日、CTD 2.6.4.5.5)
4
Teichert J. et al. :Drug Metab. Dispos. 2005; 33: 984
5
Teichert J. et al. :Drug Metab. Dispos. 2009; 37: 292
6
社内資料:薬物動態[海外臨床試験(98B03試験)]
(承認年月日:2010年10月27日、CTD 2.7.6.8)
(承認年月日:2010年10月27日、CTD 2.7.6.8)
7
Ohmachi K. et al. :Cancer Sci. 2010; 101: 2059
8
Ogura M. et al. :Int. J. Hematol. 2017; 105: 470
9
社内資料:海外第Ⅲ相臨床試験(NHL 1-2003試験)
(承認年月日:2016年12月19日、CTD 2.7.6.1)
(承認年月日:2016年12月19日、CTD 2.7.6.1)
10
Marcus R. et al. :N. Engl. J. Med. 2017; 377: 1331
11
Sehn LH. et al. :Lancet Oncol. 2016; 17: 1081
12
社内資料:国内第Ⅲ相臨床試験(2017002試験)
(承認年月日:2021年3月23日、CTD 2.7.6.2)
(承認年月日:2021年3月23日、CTD 2.7.6.2)
13
社内資料:海外第Ⅰb/Ⅱ相試験(GO29365試験)
(承認年月日:2021年3月23日、CTD2.7.3.2、2.7.4.2.1.1)
(承認年月日:2021年3月23日、CTD2.7.3.2、2.7.4.2.1.1)
14
Sehn LH, et al. J Clin Oncol 2020;38:155-65
15
社内資料:国内第Ⅱ相試験(JO40762試験)
(承認年月日:2021年3月23日、CTD2.7.3.2、2.7.4.2.1.1)
(承認年月日:2021年3月23日、CTD2.7.3.2、2.7.4.2.1.1)
16
社内資料:海外第Ⅲ相臨床試験(02CLL Ⅲ試験)
(承認年月日:2016年8月26日、CTD 2.7.6.1)
(承認年月日:2016年8月26日、CTD 2.7.6.1)
17
Strumberg D. et al. :Anticancer Drugs 1996; 7: 415
18
Leoni L.M. et al. :Clin. Cancer Res. 2008; 14: 309
19
Gaul L. et al. :J. Cancer Res. Clin. Oncol. 2008; 134: 245
20
Roue G. et al. :Clin. Cancer Res. 2008; 14: 6907
21
Alonso R. et al. :Blood 2009; 114: 1563
22
社内資料:薬効薬理[ベンダムスチンのヒト低悪性度B細胞性非ホジキンリンパ腫由来細胞株及びマントル細胞リンパ腫由来細胞株に対する細胞増殖抑制作用]
(承認年月日:2010年10月27日、CTD 2.6.2.2.1.7、CTD 2.6.2.2.2.7)
(承認年月日:2010年10月27日、CTD 2.6.2.2.1.7、CTD 2.6.2.2.2.7)
23
社内資料:薬効薬理[ベンダムスチンのヒト慢性リンパ性白血病細胞株に対する細胞増殖抑制作用]
(承認年月日:2016年8月26日、CTD 2.6.2.2.1.1、CTD 2.6.2.2.2.1)
(承認年月日:2016年8月26日、CTD 2.6.2.2.1.1、CTD 2.6.2.2.2.1)
24
社内資料:薬効薬理[ベンダムスチンのヒトびまん性大細胞型B細胞リンパ腫由来細胞株に対する細胞増殖抑制作用]
(承認年月日:2021年3月23日、CTD 2.6.2.2.1.1、CTD 2.6.2.2.2.1)
(承認年月日:2021年3月23日、CTD 2.6.2.2.1.1、CTD 2.6.2.2.2.1)

