


1
Fisher CJ, et al.:N Engl J Med. 1996;334(26):1697-1702
2
Mann DL, et al.:Circulation. 2004;109(13):1594-1602
3
Chung ES, et al.:Circulation. 2003;107(25):3133-3140
4
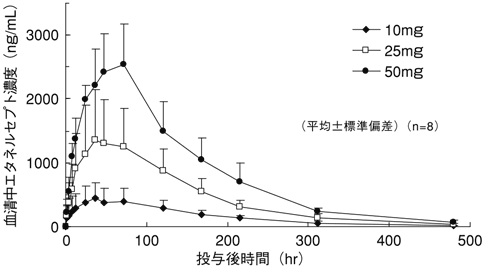
Kawai S, et al.:J Clin Pharmacol. 2006;46(4):418-423
5
Korth-Bradley J, et al.:Ann Pharmacother. 2000;34(2):161-164
6
社内資料:生物学的同等性試験結果:20021643試験
7
Sullivan JT, et al.:J Clin Pharmacol. 2006;46(6):654-661
8
Takeuchi T, et al.:Mod Rheumatol. 2015;25(2):173-186
9
Takeuchi T, et al.:Mod Rheumatol. 2013;23(4):623-633
10
Moreland LW, et al.:N Engl J Med. 1997;337(3):141-147
11
Moreland LW, et al.:Ann Intern Med. 1999;130(6):478-486
12
Genovese MC, et al.:Arthritis Rheum. 2002;46(6):1443-1450

