





1
社内資料: ラットにおける生殖発生毒性試験(2008年1月25日承認、CTD2.6.6.6.1)
2
社内資料: ウサギにおける生殖発生毒性試験(2008年1月25日承認、CTD2.6.6.6.2)
3
社内資料: イヌにおける反復投与毒性試験(2008年1月25日承認、CTD2.6.6.3.2.1)
4
Mross K, et al.: Eur J Cancer. 2007; 43: 55-63
5
社内資料: ドキソルビシンとの相互作用(2008年1月25日承認、CTD2.7.2.2.4.3.3)
6
社内資料: リファンピシンとの相互作用
7
社内資料: ワルファリンとの相互作用(2008年1月25日承認、CTD2.7.2.3.4.2.2)
8
社内資料: ドセタキセルとの相互作用
9
社内資料: フラジオマイシン(ネオマイシン)との相互作用(2014年6月20日承認、CTD2.7.6.14)
10
NDBを用いた調査結果の概要(VEGF/VEGFR阻害作用を有する薬剤の動脈解離に関するリスク評価):
https://www.pmda.go.jp/files/000266521.pdf
https://www.pmda.go.jp/files/000266521.pdf
11
社内資料: ラットにおける反復投与毒性試験(2008年1月25日承認、CTD2.6.6.3.1.1)
12
社内資料: ラットにおける反復投与毒性試験(2008年1月25日承認、CTD2.6.6.3.1.2)
13
社内資料: イヌにおける反復投与毒性試験(2008年1月25日承認、CTD2.6.6.3.2.3)
14
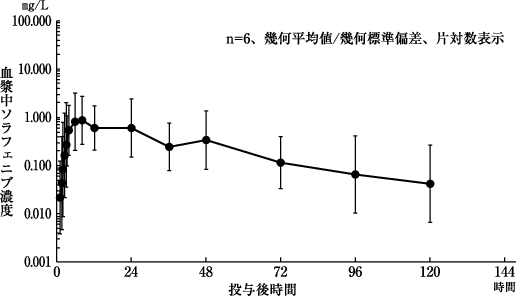
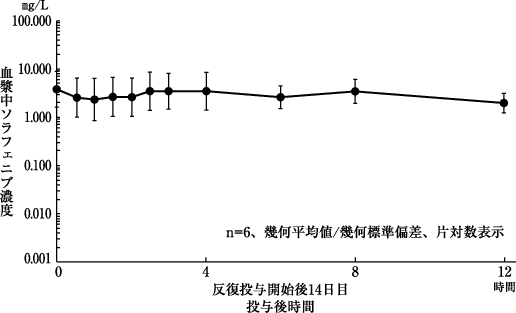
社内資料: 薬物動態(2008年1月25日承認、CTD2.7.2.2.1)
15
社内資料: 食事の影響(外国人)(2008年1月25日承認、CTD2.7.1.2.3)
16
社内資料: タンパク結合(2008年1月25日承認、CTD2.6.4.4.1)
17
社内資料: 薬物動態(外国人)(2008年1月25日承認、CTD2.7.2.2.3.1)
18
社内資料: 腎機能障害のある被験者における薬物動態(外国人)
19
Furuse J, et al.: Cancer Sci. 2008; 99: 159-165
20
社内資料: 薬物間相互作用(in vitro)(2008年1月25日承認、CTD2.6.4.5.1.5)
21
Lathia C, et al.: Cancer Chemother Pharmacol. 2006; 57: 685-692
22
社内資料: 薬物間相互作用(外国人)(2008年1月25日承認、CTD2.7.2.2.4.2)
23
社内資料: 腎細胞癌患者を対象とした第Ⅱ相臨床試験(2008年1月25日承認、CTD2.7.6.23)
24
Escudier B, et al.: N Engl J Med. 2007; 356: 125-134
25
Llovet JM, et al.: N Engl J Med. 2008; 359: 378-390
26
社内資料: 甲状腺癌患者を対象とした第Ⅲ相臨床試験(2014年6月20日承認、CTD2.7.6.1)
27
社内資料: 甲状腺未分化癌及び甲状腺髄様癌患者を対象とした第Ⅱ相臨床試験
28
Kudo M, et al.: Eur J Cancer. 2011; 47: 2117-2127
29
社内資料: 肝細胞癌患者を対象とした市販後の国際共同第Ⅲ相臨床試験
30
Wilhelm SM, et al.: Cancer Research. 2004; 64: 7099-7109
31
Chang YS, et al.: Cancer Chemother Pharmacol. 2007; 59: 561-574
32
Liu L, et al.: Cancer Research. 2006; 66: 11851-11858

